
计算物理 ›› 2023, Vol. 40 ›› Issue (1): 40-46.DOI: 10.19596/j.cnki.1001-246x.8543
程翔1( ), 冯军伟2, EvgeniiTikhonov3
), 冯军伟2, EvgeniiTikhonov3
收稿日期:2022-04-12
出版日期:2023-01-25
发布日期:2023-07-04
作者简介:程翔(1992-),男,江西德兴,助教,研究方向为基于第一性原理的材料计算,E-mail: cx1553596515@126.com
基金资助:
Xiang CHENG1(), Junwei FENG2, Tikhonov EVGENII3
Received:2022-04-12
Online:2023-01-25
Published:2023-07-04
摘要:
基于进化算法和第一性原理计算,预测四元化合物HfxTa8-xC7N(x=1~7)的晶体结构。与二元HfC和TaC的晶体结构相似,这些四元HfxTa8-xC7N晶体结构属于岩盐结构。计算结果表明:随着有效价电子浓度(VEC)的增大,HfxTa8-xC7N的体模量逐渐增大;当VEC等于8.875时,剪切模量和杨氏模量达到最大值;维氏硬度在VEC等于8.25时达到最大值。因此,可以通过VEC的设计,得到具有综合力学性质优异的四元HfxTa8-xC7N化合物。计算HfxTa8-xC7N的电子性质,并分析力学性质与电子性质之间的关系。
程翔, 冯军伟, EvgeniiTikhonov. Hf-Ta-C-N四元化合物的结构、力学及电子性质的第一性原理研究[J]. 计算物理, 2023, 40(1): 40-46.
Xiang CHENG, Junwei FENG, Tikhonov EVGENII. First-principles Study of Structures, Mechanical Properties and Electronic Properties of Quaternary Hf-Ta-C-N System[J]. Chinese Journal of Computational Physics, 2023, 40(1): 40-46.
| Compound | Space group | Lattice constants/Å | ΔHf/(eV·atom-1) | instability/(eV·atom-1) | VEC |
| Hf7TaC7N | P4mm | a=4.603, b=4.603, c=9.245 | -1.041 5 | 0.004 1 | 8.25 |
| Hf6Ta2C7N | Pmmm | a=4.575, b=4.600, c=9.166 | -0.993 7 | 0.010 7 | 8.375 |
| Hf5Ta3C7N | Pmm2 | a=4.560, b=4.564, c=9.145 | -0.945 1 | 0.013 3 | 8.5 |
| Hf4Ta4C7N | Pmm2 | a=4.538, b=4.548, c=9.094 | -0.897 5 | 0.007 1 | 8.625 |
| Hf3Ta5C7N | P4mm | a=4.515, b=4.515, c=9.091 | -0.842 4 | 0.005 9 | 8.75 |
| Hf2Ta6C7N | Pmm2 | a=4.495, b=4.503, c=9.030 | -0.781 2 | 0.007 9 | 8.875 |
| HfTa7C7N | P4mm | a=4.488, b=4.488, c=8.959 | -0.702 8 | 0.022 6 | 9 |
表1 HfxTa8-xC7N(x=1~7)的空间群、晶格常数、形成焓ΔHf、高出凸包线的能量和有效价电子浓度(VEC)
Table 1 Space groups, lattice constants, enthalpy of formation ΔHf, enthalpy above convex-hull, and valence electron concentrations (VEC) of HfxTa8-xC7N (x=1-7)
| Compound | Space group | Lattice constants/Å | ΔHf/(eV·atom-1) | instability/(eV·atom-1) | VEC |
| Hf7TaC7N | P4mm | a=4.603, b=4.603, c=9.245 | -1.041 5 | 0.004 1 | 8.25 |
| Hf6Ta2C7N | Pmmm | a=4.575, b=4.600, c=9.166 | -0.993 7 | 0.010 7 | 8.375 |
| Hf5Ta3C7N | Pmm2 | a=4.560, b=4.564, c=9.145 | -0.945 1 | 0.013 3 | 8.5 |
| Hf4Ta4C7N | Pmm2 | a=4.538, b=4.548, c=9.094 | -0.897 5 | 0.007 1 | 8.625 |
| Hf3Ta5C7N | P4mm | a=4.515, b=4.515, c=9.091 | -0.842 4 | 0.005 9 | 8.75 |
| Hf2Ta6C7N | Pmm2 | a=4.495, b=4.503, c=9.030 | -0.781 2 | 0.007 9 | 8.875 |
| HfTa7C7N | P4mm | a=4.488, b=4.488, c=8.959 | -0.702 8 | 0.022 6 | 9 |
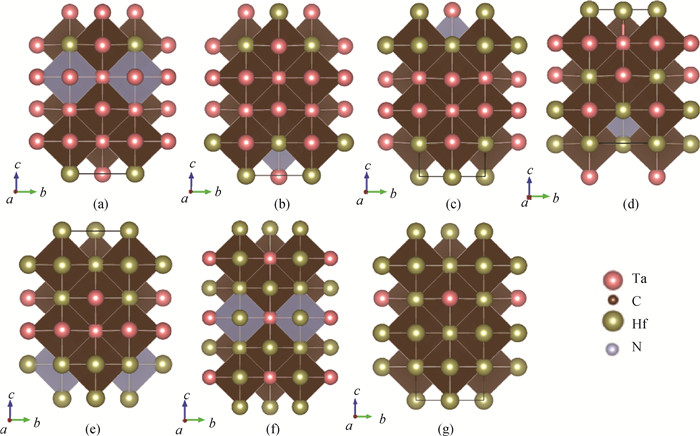
图1 晶体结构(a)HfTa7C7N;(b)Hf2Ta6C7N;(c)Hf3Ta5C7N;(d)Hf4Ta4C7N;(e)Hf5Ta3C7N;(f)Hf6Ta2C7N;(g)Hf7TaC7N
Fig.1 Crystal structures of (a) HfTa7C7N, (b) Hf2Ta6C7N, (c) Hf3Ta5C7N, (d) Hf4Ta4C7N, (e) Hf5Ta3C7N, (f) Hf6Ta2C7N and (g) Hf7TaC7N
| Compound | Bond length/Å | Volume per atom/Å3 | |||
| Hf-C | Hf-N | Ta-C | Ta-N | ||
| Hf7TaC7N | 2.285~2.328 | 2.302~2.364 | 2.199~2.303 | 2.353 | 12.243 5 |
| Hf6Ta2C7N | 2.273~2.309 | 2.288~2.306 | 2.259~2.323 | 2.300 | 12.055 2 |
| Hf5Ta3C7N | 2.273~2.349 | 2.280~2.373 | 2.164~2.301 | 2.335 | 11.892 5 |
| Hf4Ta4C7N | 2.233~2.289 | 2.269~2.344 | 2.158~2.301 | 2.274~2.359 | 11.731 4 |
| Hf3Ta5C7N | 2.258~2.322 | 2.258 | 2.163~2.259 | 2.352~2.413 | 11.583 0 |
| Hf2Ta6C7N | 2.248~2.285 | 2.248 | 2.178~2.272 | 2.251~2.349 | 11.423 4 |
| HfTa7C7N | 2.238~2.244 | 2.272 | 2.184~2.275 | 2.244~2.264 | 11.275 4 |
| Hf8C7N | 2.309~2.322 | 3.324 | 12.432 8 | ||
表2 HfxTa8-xC7N(x=1~8)的键长和原子体积
Table 2 Bond lengths and volume per atom of HfxTa8-xC7N(x=1-8)
| Compound | Bond length/Å | Volume per atom/Å3 | |||
| Hf-C | Hf-N | Ta-C | Ta-N | ||
| Hf7TaC7N | 2.285~2.328 | 2.302~2.364 | 2.199~2.303 | 2.353 | 12.243 5 |
| Hf6Ta2C7N | 2.273~2.309 | 2.288~2.306 | 2.259~2.323 | 2.300 | 12.055 2 |
| Hf5Ta3C7N | 2.273~2.349 | 2.280~2.373 | 2.164~2.301 | 2.335 | 11.892 5 |
| Hf4Ta4C7N | 2.233~2.289 | 2.269~2.344 | 2.158~2.301 | 2.274~2.359 | 11.731 4 |
| Hf3Ta5C7N | 2.258~2.322 | 2.258 | 2.163~2.259 | 2.352~2.413 | 11.583 0 |
| Hf2Ta6C7N | 2.248~2.285 | 2.248 | 2.178~2.272 | 2.251~2.349 | 11.423 4 |
| HfTa7C7N | 2.238~2.244 | 2.272 | 2.184~2.275 | 2.244~2.264 | 11.275 4 |
| Hf8C7N | 2.309~2.322 | 3.324 | 12.432 8 | ||
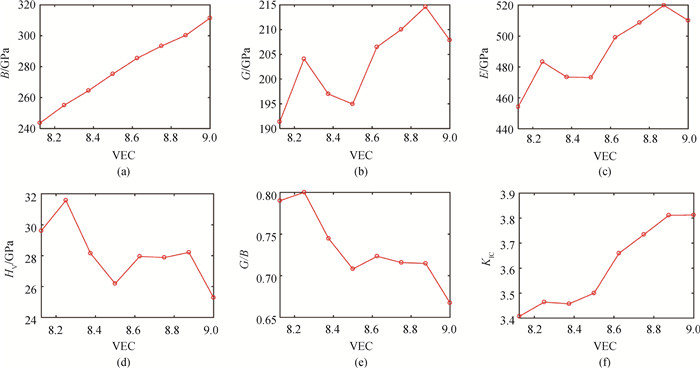
图2 HfxTa8-xC7N (x=1~8)的力学性质与VEC之间的关系(a) 体模量(B);(b) 剪切模量(G);(c) 杨氏模量(E);(d) 维氏硬度(HV);(e) Pugh比(G/B);(f) 断裂韧性(KIC)
Fig.2 Mechanical properties as functions of VEC for HfxTa8-xC7N (x=1-8) (a) Bulk modulus (B); (b) shear modulus (G); (c) elastic modulus (E); (d) Vickers hardness (HV); (e) Pugh's ratio (G/B) and (f) fracture toughness (KIC)
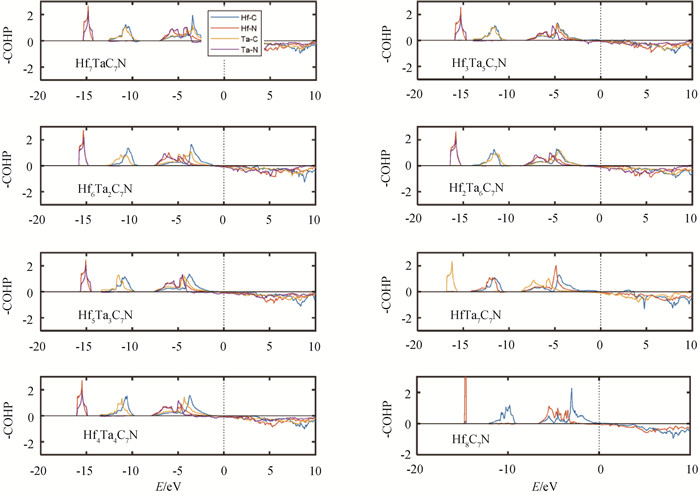
图3 HfxTa8-xC7N (x=1~8)的晶体轨道哈密顿分布(-COHP)(Fermi能级位于0 eV。)
Fig.3 Crystal orbital Hamilton populations (-COHP) of HfxTa8-xC7N (x=1-8) (The Fermi level is set at 0 eV.)
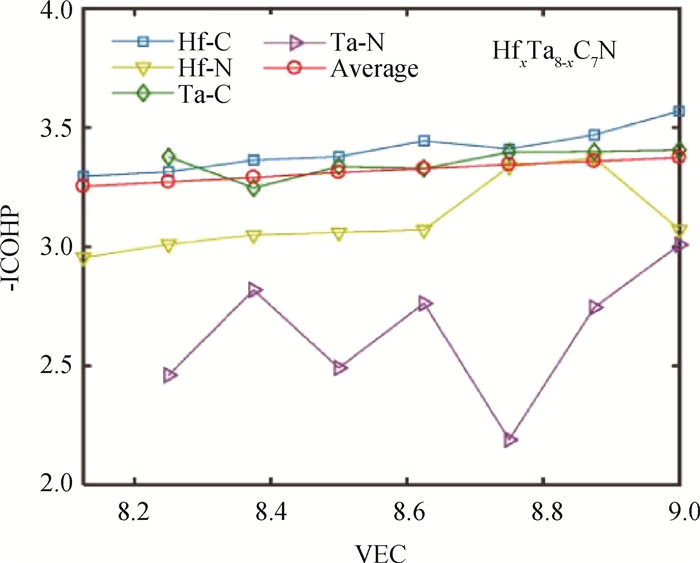
图4 HfxTa8-xC7N (x=1~8)的晶体轨道哈密顿分布积分值(-ICOHP)与VEC之间的关系
Fig.4 Integrated crystal orbital Hamilton populations (-ICOHP)as a function of VEC for HfxTa8-xC7N (x=1-8)
| 1 |
|
| 2 |
DOI |
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
DOI |
| 7 |
DOI |
| 8 |
|
| 9 |
DOI |
| 10 |
DOI |
| 11 |
DOI |
| 12 |
DOI |
| 13 |
DOI |
| 14 |
DOI |
| 15 |
DOI |
| 16 |
DOI |
| 17 |
|
| 18 |
DOI |
| 19 |
DOI |
| 20 |
DOI |
| 21 |
DOI |
| 22 |
DOI |
| 23 |
DOI |
| 24 |
DOI |
| 25 |
|
| 26 |
|
| 27 |
|
| [1] | 潘靖, 沈国华. 等价阴-阳离子共掺杂调节ZnO的能带结构及其光催化活性[J]. 计算物理, 2021, 38(3): 371-378. |
| [2] | 赵一程, 郭俊宏, 胡芳仁. 应变对单层砷烯结构拉曼散射的影响[J]. 计算物理, 2020, 37(3): 365-370. |
| [3] | 熊宗刚, 杜娟, 张现周. 二维GeSe纳米片五族和七族原子掺杂的受主和施主杂质态[J]. 计算物理, 2019, 36(6): 733-741. |
| [4] | 李永, 李海生, 李冠亚, 王赵武, 李国岭, 左正伟, 李立本. 合金效应加强水分子在PtRun团簇上的吸附作用[J]. 计算物理, 2017, 34(2): 230-236. |
| [5] | 张若兴, 侯士敏, 丑强. 基于第一性原理量子输运模拟的并行计算[J]. 计算物理, 2015, 32(6): 631-638. |
| [6] | 陈金繁, 罗超, 敖冰云, 彭丽霞, 石洁. V-Ta合金力学行为的计算与实验研究[J]. 计算物理, 2014, 31(5): 609-616. |
| [7] | 李祥然, 李丹, 王春雷, 牛原, 赵红敏, 梁春军. F原子吸附TiO2:Mn(001)稀磁半导体薄膜电子结构和磁性的第一性原理计算[J]. 计算物理, 2014, 31(1): 96-102. |
| [8] | 李磊, 李丹, 刘世勇, 赵翼. Mn掺杂的ZnS(001)表面的电子态特性[J]. 计算物理, 2010, 27(2): 293-298. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
版权所有 © 《计算物理》编辑部
地址:北京市海淀区丰豪东路2号 邮编:100094 E-mail:jswl@iapcm.ac.cn
本系统由北京玛格泰克科技发展有限公司设计开发