
Chinese Journal of Computational Physics ›› 2023, Vol. 40 ›› Issue (4): 425-435.DOI: 10.19596/j.cnki.1001-246x.8594
Previous Articles Next Articles
Zhaozhao WEI( ), Kai LIU, Huijun LI
), Kai LIU, Huijun LI
Received:2022-07-17
Online:2023-07-25
Published:2023-10-13
Zhaozhao WEI, Kai LIU, Huijun LI. Molecular Dynamics Simulation of Deformation Behavior of NiAl Nanowire Under Bending[J]. Chinese Journal of Computational Physics, 2023, 40(4): 425-435.
Add to citation manager EndNote|Ris|BibTeX
URL: http://www.cjcp.org.cn/EN/10.19596/j.cnki.1001-246x.8594
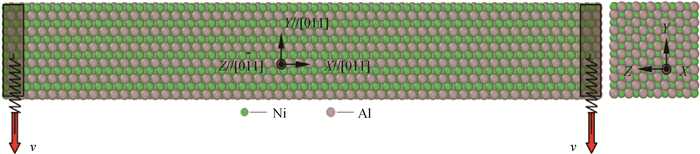
Fig.1 Initial atomic configuration of the NiAl nanowire and its square cross-section

Fig.2 F-d curve of NiAl nanowire under bending at 300 K and the evolution of fractional phase content during the deformation
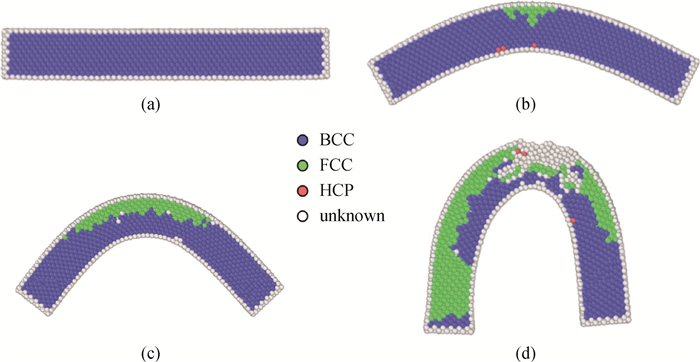
Fig.3 The corresponding atomic image of NiAl nanowire at (a) 0 ps, (b) 330 ps, (c) 560 ps and (d) 880 ps under bending at 300 K
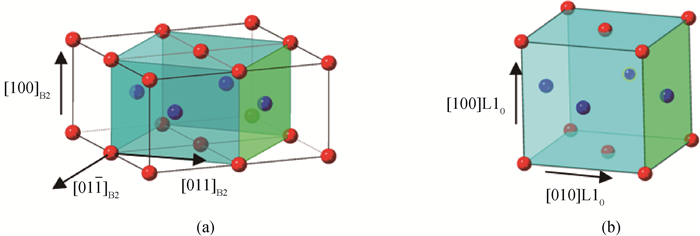
Fig.4 Crystallography of transformation from (a) B2 structure to (b) L10 structure in NiAl alloy
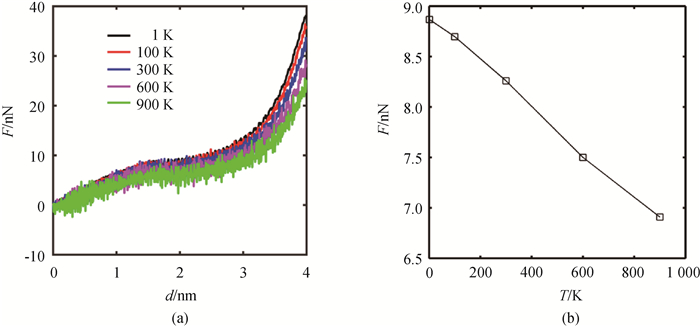
Fig.5 (a) F-d curves and (b) critical force for martensitic transformation of NiAl nanowire under bending at different temperatures
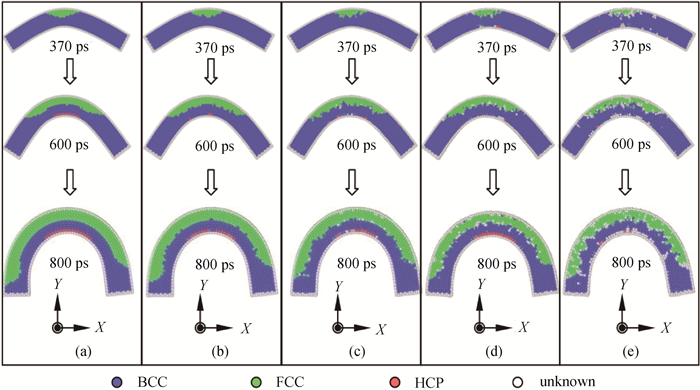
Fig.6 The corresponding atomic image of NiAl nanowire under bending at different temperatures of (a) 1 K, (b) 100 K, (c) 300 K, (d) 600 K and (e) 900 K, where colors denote the different local crystal structures: blue-BCC, green-FCC, red-HCP and white-unknown
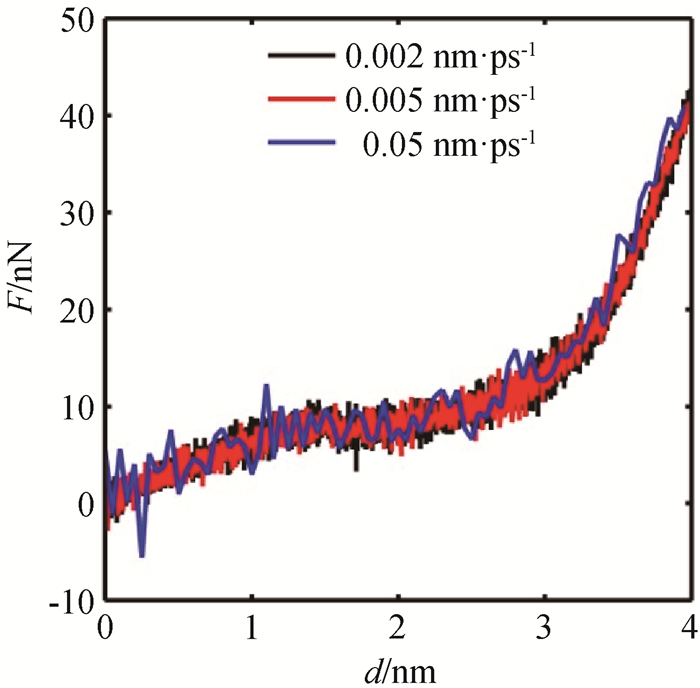
Fig.7 F-d curves of NiAl nanowire under bending at different strain rates
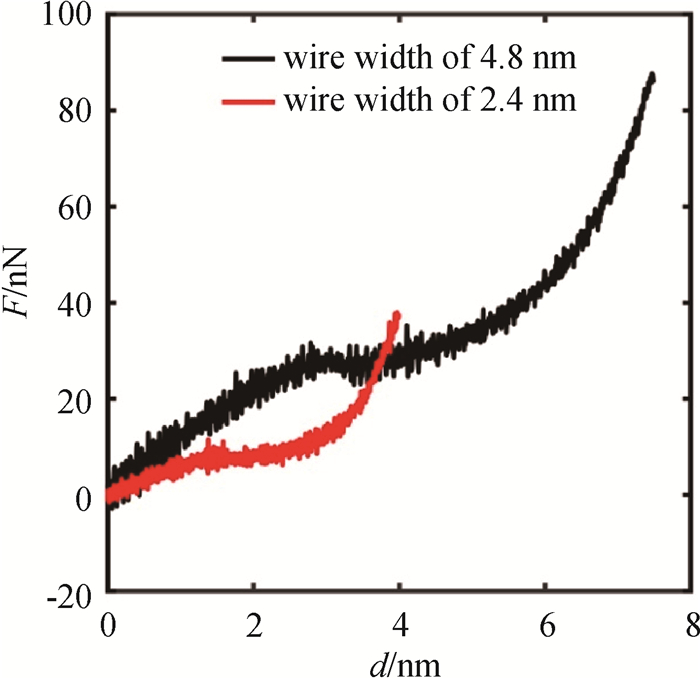
Fig.8 F-d curves of NiAl nanowires with different sizes under bending at 300 K
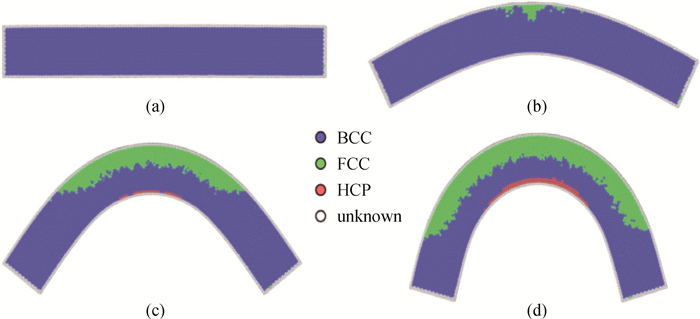
Fig.9 The corresponding atomic image at (a) 0 ps, (b) 626 ps, (c) 1 200 ps, (d) 1 500 ps of NiAl nanowire with square cross-section of larger size under bending at 300 K, where colors denote the different local crystal structures: blue-BCC, green-FCC, red-HCP and white-unknown
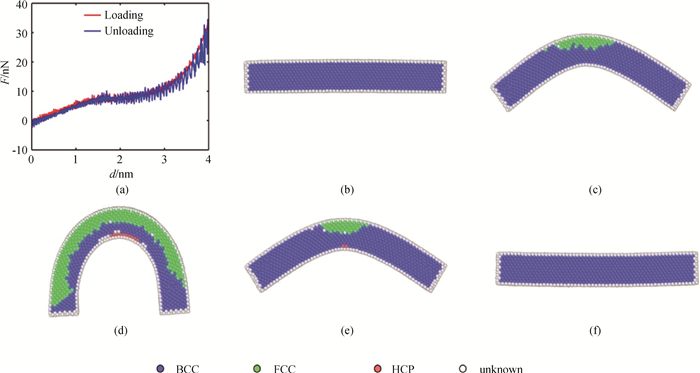
Fig.10 (a) F-d curve and the corresponding atomic images at (b) 0 ps, (c) 400 ps, (d) 800 ps, (e) 1 200 ps and (f) 1 600 ps upon loading and unloading of bending at 300 K for NiAl nanowire, where colors denote the different local crystal structures: blue-BCC, green-FCC, red- HCP and white- unknown.
| 1 |
DOI |
| 2 |
DOI |
| 3 |
DOI |
| 4 |
DOI |
| 5 |
|
| 6 |
DOI |
| 7 |
|
| 8 |
DOI |
| 9 |
DOI |
| 10 |
DOI |
| 11 |
DOI |
| 12 |
|
| 13 |
DOI |
| 14 |
DOI |
| 15 |
DOI |
| 16 |
|
| 17 |
DOI |
| 18 |
DOI |
| 19 |
DOI |
| 20 |
DOI |
| 21 |
DOI |
| 22 |
DOI |
| 23 |
DOI |
| 24 |
|
| 25 |
DOI |
| 26 |
DOI |
| 27 |
刘宁, 胡振东. 温度对NiAl合金纳米线应力诱发相变的影响[J]. 材料科学与工程学报, 2016, 34 (4): 545- 549.
DOI |
| 28 |
DOI |
| 29 |
DOI |
| 30 |
DOI |
| 31 |
DOI |
| 32 |
DOI |
| 33 |
DOI |
| 34 |
DOI |
| 35 |
DOI |
| 36 |
DOI |
| 37 |
DOI |
| 38 |
DOI |
| 39 |
DOI |
| 40 |
DOI |
| 41 |
|
| 42 |
|
| 43 |
DOI |
| 44 |
DOI |
| 45 |
DOI |
| 46 |
DOI |
| 47 |
DOI |
| 48 |
DOI |
| 49 |
DOI |
| 50 |
DOI |
| [1] | Yu LI, Huiqing LIU, Yabin FENG, Xiaohu DONG, Qing WANG, Bo ZHANG. Adsorption Behavior of Heavy Oil on Montmorillonite Surface by Typical Surfactant: Molecular Dynamics Simulation [J]. Chinese Journal of Computational Physics, 2023, 40(5): 583-596. |
| [2] | Zhaoyang HOU, Yuan NIU, Qixin XIAO, Zhen WANG, Qingtian DENG. Simulation of Mechanical Behavior and Deformation Mechanism of Al Nanowires Along Different Crystal Orientations [J]. Chinese Journal of Computational Physics, 2022, 39(3): 341-351. |
| [3] | Hubao A, Zhibing YANG, Ran HU, Yifeng CHEN. Molecular Dynamics Simulations of Capillary Dynamics at the Nanoscale [J]. Chinese Journal of Computational Physics, 2021, 38(5): 603-611. |
| [4] | WANG Guohua, CUI Yaru, YANG Ze, LI Xiaoming, TANG Hongliang, YANG Shufeng. Potential Function and Molecular Dynamics Simulation for FexO-SiO2-CaO-MgO-“NiO” Nickel Slag [J]. Chinese Journal of Computational Physics, 2021, 38(2): 215-223. |
| [5] | WANG Xuemei, DONG Bin, ZHU Ziliang, YANG Junsheng. Interfacial Interaction and Diffusion Properties of Functionalized CNT/Polymer Systems: Molecular Dynamics Simulations [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(5): 589-594. |
| [6] | HE Erbin, LUO Zhirong, ZHU Liuhua. Atomistic Analysis of Myoglobin Mechanical Unfolding [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(2): 205-211. |
| [7] | ZHOU Lu, MA Honghe. Molecular Dynamics Simulation on Crystallization Kinetics of Sodium Sulfate in Supercritical Water [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(2): 212-220. |
| [8] | CHAI Rukuan, LIU Yuetian, WANG Junqiang, XIN Jing, PI Jian, LI Changyong. Molecular Dynamics Simulation of Wettability of Calcite and Dolomite [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(4): 474-482. |
| [9] | WANG Shuaichuang, ZHANG Gongmu, SUN Bo, SONG Haifeng, TIAN Mingfeng, FANG Jun, LIU Haifeng. Quantum Molecular Dynamics Simulations of Transport Properties of Liquid Plutonium [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(3): 253-258. |
| [10] | LIANG Hua, LI Maosheng. Molecular Dynamics Study of Mechanical Properties of Single Crystal Aluminum with Voids and Vacancies [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(2): 211-218. |
| [11] | ZHANG Haiyan, YIN Xinchun. Molecular Dynamics Study on Growth Mechanism of Pure Metals Solid-Liquid Interface During Solidification [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(1): 80-88. |
| [12] | QI Meiling, YANG Qiong, WANG Canglong, TIAN Yuan, YANG Lei. Parallel Algorithm on GPU and Optimization for Molecular Dynamics Simulation of Irradiation Damage of Structure Materials [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2017, 34(4): 461-467. |
| [13] | TIAN Wenchao, CHEN Zhiqiang, SHAN Lei. Numerical Analysis and Simulation of a Low-voltage Large-displacement Micro-actuator [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2014, 31(2): 223-229. |
| [14] | LIU Bing, SHI Junqin, SHEN Yue, ZHANG Jun. A Molecular Dynamics Simulation of Methane Adsorption in Graphite Slit-pores [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2013, 30(5): 692-699. |
| [15] | CHEN Shenshen, LI Qinghua, OU Manli. Meshless Kriging Interpolation Method for Bending of a Moderately Thick Plate [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2013, 30(4): 547-553. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
Copyright © Chinese Journal of Computational Physics
E-mail: jswl@iapcm.ac.cn
Supported by Beijing Magtech Co., Ltd.