
Chinese Journal of Computational Physics ›› 2021, Vol. 38 ›› Issue (4): 447-455.DOI: 10.19596/j.cnki.1001-246x.8263
• Research Reports • Previous Articles Next Articles
Yuxing YAN1( ), Juexuan ZHANG1, Shuai ZHENG2, Fan WANG1, Linqiang XIONG3
), Juexuan ZHANG1, Shuai ZHENG2, Fan WANG1, Linqiang XIONG3
Received:2020-08-24
Online:2021-07-25
Published:2021-12-21
CLC Number:
Yuxing YAN, Juexuan ZHANG, Shuai ZHENG, Fan WANG, Linqiang XIONG. First-principles Study of Electronic Structure and Optical Properties of ZnNb2O6 with Interstitial Atoms[J]. Chinese Journal of Computational Physics, 2021, 38(4): 447-455.
Add to citation manager EndNote|Ris|BibTeX
URL: http://www.cjcp.org.cn/EN/10.19596/j.cnki.1001-246x.8263
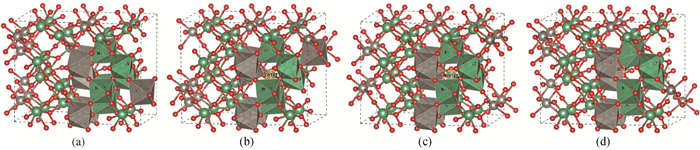
Fig.1 Crystal structure of (a) ZnNb2O6, (b) Zni, (c) Nbi and (d) Oi (The gray, green and red are Zn, Nb and O atoms, respectively.)

Fig.2 Influence of functional on (a) energy gap and (b) lattice length
| Epure/eV | Edefect/eV | Eform/eV | |
| ZnNb2O6 | -7 998.113 | ||
| Zni | -7 859.723 | 8.433 | |
| Nbi | -7 851.647 | 5.289 | |
| Oi | -7 775.457 | 7.495 |
Table 1 Total energies and formation energies of ZnNb2O6 with different interstitial atoms
| Epure/eV | Edefect/eV | Eform/eV | |
| ZnNb2O6 | -7 998.113 | ||
| Zni | -7 859.723 | 8.433 | |
| Nbi | -7 851.647 | 5.289 | |
| Oi | -7 775.457 | 7.495 |
| Model | a/nm | b/nm | c/nm | α | β | γ | ΔV/V/% |
| ZnNb2O6(Ref.[ | 1.420 8 | 0.572 6 | 0.504 0 | 90 | 90 | 90 | |
| ZnNb2O6(This work) | 1.433 4 | 0.583 2 | 0.506 1 | 90.000 1 | 90.000 8 | 90.000 2 | 3.18 |
| Zni | 1.440 4 | 0.584 6 | 0.507 8 | 90.047 1 | 89.604 3 | 90.281 0 | 1.086 |
| Nbi | 1.440 1 | 0.585 0 | 0.507 3 | 89.897 2 | 89.910 6 | 89.721 1 | 1.011 |
| Oi | 1.436 5 | 0.584 0 | 0.507 1 | 89.722 9 | 90.127 9 | 90.036 5 | 0.541 |
Table 2 Lattice parameters of the model systems
| Model | a/nm | b/nm | c/nm | α | β | γ | ΔV/V/% |
| ZnNb2O6(Ref.[ | 1.420 8 | 0.572 6 | 0.504 0 | 90 | 90 | 90 | |
| ZnNb2O6(This work) | 1.433 4 | 0.583 2 | 0.506 1 | 90.000 1 | 90.000 8 | 90.000 2 | 3.18 |
| Zni | 1.440 4 | 0.584 6 | 0.507 8 | 90.047 1 | 89.604 3 | 90.281 0 | 1.086 |
| Nbi | 1.440 1 | 0.585 0 | 0.507 3 | 89.897 2 | 89.910 6 | 89.721 1 | 1.011 |
| Oi | 1.436 5 | 0.584 0 | 0.507 1 | 89.722 9 | 90.127 9 | 90.036 5 | 0.541 |
| Model | Bond | Population min(max) | Length min(max)/nm |
| ZnNb2O6 | O-Zn | 0.15(0.29) | 0.207 222(0.222 210 0) |
| O-Nb | 0.21(0.79) | 0.181 660(0.233 858) | |
| O-O | -0.05(0.00) | 0.262 644(0.292 990) | |
| Zni | O-Zn | 0.04(0.43) | 0.193 899(0.264 244) |
| O-Nb | 0.09(0.83) | 0.180 471(0.253 899) | |
| O-O | -0.05(0.00) | 0.260 592(0.299 858) | |
| Zn-Nb | -0.48(-0.30) | 0.284 044(0.299 528) | |
| Zn-Zn | -1.87 | 0.264 037 | |
| Nb-Nb | -0.45 | 0.273 866 | |
| Nbi | O-Zn | -0.01(0.39) | 0.196 825(0.254 825) |
| O-Nb | 0.10(0.82) | 0.180 989(0.245 336) | |
| O-O | -0.05(0.00) | 0.259 177(0.299 995) | |
| Zn-Nb | 0.18 | 0.279 478 | |
| Zn-Zn | |||
| Nb-Nb | -0.81(-0.41) | 0.267 331(0.288 840) | |
| Oi | O-Zn | -0.08(0.36) | 0.195 009(0.288 686) |
| O-Nb | 0.04(0.81) | 0.180 078(0.292 940) | |
| O-O | -0.06(0.18) | 0.145 112(0.298 924) |
Table 3 Bond populations of the model systems
| Model | Bond | Population min(max) | Length min(max)/nm |
| ZnNb2O6 | O-Zn | 0.15(0.29) | 0.207 222(0.222 210 0) |
| O-Nb | 0.21(0.79) | 0.181 660(0.233 858) | |
| O-O | -0.05(0.00) | 0.262 644(0.292 990) | |
| Zni | O-Zn | 0.04(0.43) | 0.193 899(0.264 244) |
| O-Nb | 0.09(0.83) | 0.180 471(0.253 899) | |
| O-O | -0.05(0.00) | 0.260 592(0.299 858) | |
| Zn-Nb | -0.48(-0.30) | 0.284 044(0.299 528) | |
| Zn-Zn | -1.87 | 0.264 037 | |
| Nb-Nb | -0.45 | 0.273 866 | |
| Nbi | O-Zn | -0.01(0.39) | 0.196 825(0.254 825) |
| O-Nb | 0.10(0.82) | 0.180 989(0.245 336) | |
| O-O | -0.05(0.00) | 0.259 177(0.299 995) | |
| Zn-Nb | 0.18 | 0.279 478 | |
| Zn-Zn | |||
| Nb-Nb | -0.81(-0.41) | 0.267 331(0.288 840) | |
| Oi | O-Zn | -0.08(0.36) | 0.195 009(0.288 686) |
| O-Nb | 0.04(0.81) | 0.180 078(0.292 940) | |
| O-O | -0.06(0.18) | 0.145 112(0.298 924) |
| Model | Species | s | p | d | Total | Charge/eV |
| Zn | 0.11 | 0.62 | 9.98 | 10.72 | 1.28 | |
| ZnNb2O6 | Nb | 2.35 | 6.31 | 2.92 | 11.58 | 1.42 |
| O | 1.84~1.86 | 4.80~4.86 | 6.67~6.70 | -(0.70~0.67) | ||
| Zn | 0.07~0.16 | 0.62~0.81 | 9.97~9.98 | 10.68~10.87 | 1.13~1.32 | |
| Zni | Nb | 2.35~2.37 | 6.29~6.34 | 2.91~3.17 | 11.59~11.81 | 1.19~1.41 |
| O | 1.84~1.86 | 4.79~4.93 | 6.65~6.74 | -(0.78~0.65) | ||
| Zn | 0.06~0.10 | 0.61~0.64 | 9.98 | 10.67~10.70 | 1.30~1.33 | |
| Nbi | Nb | 2.34~2.36 | 6.24~6.34 | 2.91~3.65 | 11.58~12.31 | 0.69~1.42 |
| O | 1.84~1.86 | 4.79~4.87 | 6.66~6.72 | -(0.71~0.66) | ||
| Zn | 0.06~0.07 | 0.61~0.63 | 9.98 | 10.66~10.68 | 1.32~1.34 | |
| Oi | Nb | 2.35~2.36 | 6.30~6.34 | 2.88~2.93 | 11.54~11.61 | 1.39~1.46 |
| O | 1.84~1.90 | 4.50~4.87 | 6.39~6.71 | -(0.71~0.39) |
Table 4 Atomic populations of the model systems
| Model | Species | s | p | d | Total | Charge/eV |
| Zn | 0.11 | 0.62 | 9.98 | 10.72 | 1.28 | |
| ZnNb2O6 | Nb | 2.35 | 6.31 | 2.92 | 11.58 | 1.42 |
| O | 1.84~1.86 | 4.80~4.86 | 6.67~6.70 | -(0.70~0.67) | ||
| Zn | 0.07~0.16 | 0.62~0.81 | 9.97~9.98 | 10.68~10.87 | 1.13~1.32 | |
| Zni | Nb | 2.35~2.37 | 6.29~6.34 | 2.91~3.17 | 11.59~11.81 | 1.19~1.41 |
| O | 1.84~1.86 | 4.79~4.93 | 6.65~6.74 | -(0.78~0.65) | ||
| Zn | 0.06~0.10 | 0.61~0.64 | 9.98 | 10.67~10.70 | 1.30~1.33 | |
| Nbi | Nb | 2.34~2.36 | 6.24~6.34 | 2.91~3.65 | 11.58~12.31 | 0.69~1.42 |
| O | 1.84~1.86 | 4.79~4.87 | 6.66~6.72 | -(0.71~0.66) | ||
| Zn | 0.06~0.07 | 0.61~0.63 | 9.98 | 10.66~10.68 | 1.32~1.34 | |
| Oi | Nb | 2.35~2.36 | 6.30~6.34 | 2.88~2.93 | 11.54~11.61 | 1.39~1.46 |
| O | 1.84~1.90 | 4.50~4.87 | 6.39~6.71 | -(0.71~0.39) |
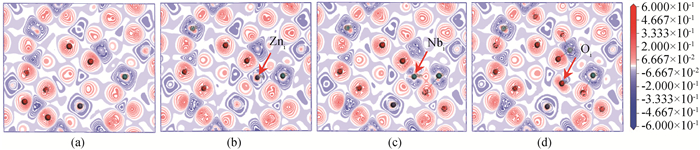
Fig.3 Electron density difference interface of (a) ZnNb2O6, (b) Zni, (c) Nbi and (d) Oi
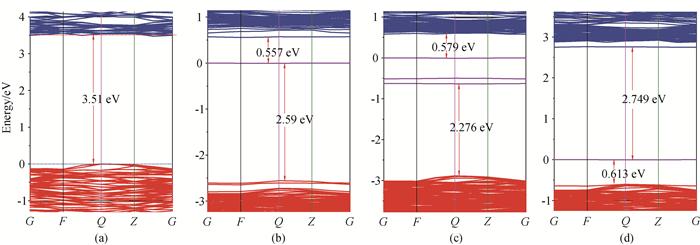
Fig.4 Band structures of the model systems (a) ZnNb2O6, (b) Zni, (c) Nbi and (d) Oi

Fig.5 Total and partial densities of states of the model systems (a) ZnNb2O6, (b) Zni, (c) Nbi and (d) Oi
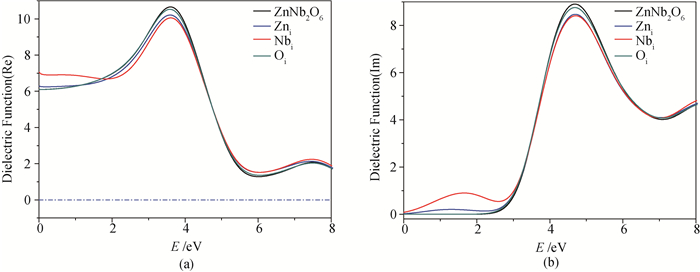
Fig.6 Dielectric functions of the model systems (a) real part and (b) imaginary part
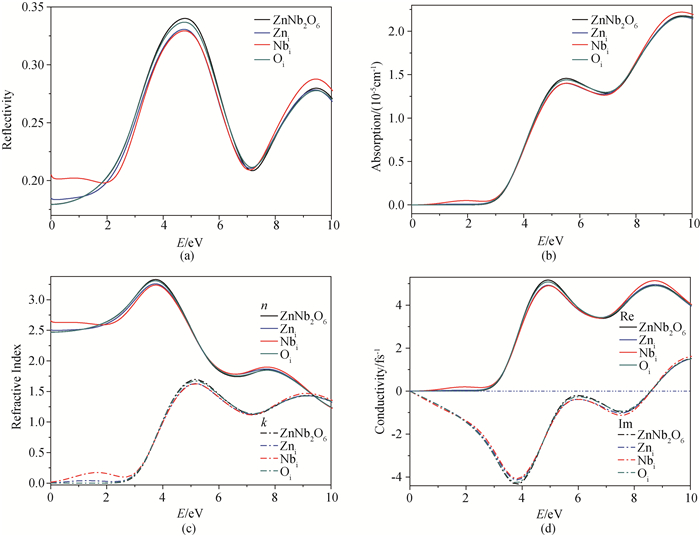
Fig.7 Photoelectric characteristic functions of the model systems (a) reflectivity, (b) absorption, (c) refractive index, (d) conductivity
| 1 |
LIU C, LI F, MA L P, et al. Advanced materials for energy storage[J]. Advanced Materials, 2010, 22 (8): E28- E62.
DOI |
| 2 |
CABALLER A C, FERNABDEZ J F, MOURE C, et al. ZnO-doped BaTiO3: Microstructure and electrical properties[J]. Journal of the European Ceramic Society, 1997, 17 (4): 513- 523.
DOI |
| 3 |
DONG G X, MA S W, DU J, et al. Dielectric properties and energy storage density in ZnO-doped Ba0.3Sr0.7TiO3 ceramics[J]. Ceramics International, 2009, 35 (5): 2069- 2075.
DOI |
| 4 |
ZHANG Y C, YUE Z X, GUI Z L, et al. Effects of CaF2 addition on the microstructure and microwave dielectric properties of ZnNb2O6 ceramics[J]. Ceramics International, 2003, 29 (5): 555- 559.
DOI |
| 5 |
NGAMJARUROJANA A, YIMNIRUN R, ANANTA S. Effect of calcination conditions on phase formation and particle size of zinc niobate powers synthesized by solid-state reaction[J]. Materials Letters, 2006, 60 (23): 2867- 2872.
DOI |
| 6 | XU D P, LIU Y, ZHOU Q, et al. Optical phonon behaviors of columbite ZnNb2O6 single crystal[J]. Journal of Alloys and Compounds, 2015, (618): 694- 699. |
| 7 | NEURGAONKAR R R, CORY W K, OLIVER J R. Growth and ferroelectric properties of tungsten bronze Sr2-xCaxNaNb5O15 single crystals[J]. Mater Res Bull, 1988, 223 (10): 1459- 1461. |
| 8 |
NEURGAONKAR R R, NELSON J G, OLIVER J R. Ferroelectric properties of tungsten bronze M62+M24-Nb8O30 solid solutions system[J]. Mater Res Bull, 1992, 27 (6): 677- 684.
DOI |
| 9 | 许煜寰. 铁电与压电材料[M]. 北京: 科学出版社, 1978: 1- 38. |
| 10 | CHEN C T, CONG S, CHEN H F, et al. First-principles study of electronic structure and optical properties of Bi doped ZnO[J]. Chinese Journal of Computational Physics, 2018, 35 (6): 720- 728. |
| 11 | 张迎春. 铌钽酸盐微波介质陶瓷材料[M]. 北京: 科学出版社, 2005: 1- 25. |
| 12 |
WU W M, LIANG S J, DING Z X, et al. A new approach to the preparation of microcrystalline ZnNb2O6 photocatalysts via a water-soluble niobium-citrate-peroxo compound[J]. Solid State Sciences, 2011, 13 (11): 2019- 2023.
DOI |
| 13 |
ZHENG J, CHEN G H, CHEN X, et al. Dielectric properties and energy storage behaviors in ZnNb2O6-doped Sr0.97Nd0.02TiO3 ceramics[J]. Journal of Materials Science: Materials in Electronics, 2016, 27 (4): 3759- 3764.
DOI |
| 14 |
WANG T, WEI X Y, HU Q Y, et al. Effects of ZnNb2O6 addition on BaTiO3 ceramics for energy storage[J]. Materials Science and Engineering B, 2013, 178 (16): 1081- 1086.
DOI |
| 15 |
ZHANG Y C, LI L T, YUE Z X, et al. Effects of additives on microstructures and microwave dielectric properties of ZnNb2O6 ceramics[J]. Materials Science and Engineering B, 2003, 99 (1-3): 282- 285.
DOI |
| 16 | 闫宇星, 汪帆, 李付绍, 等. ZnNb2-xTaxO6(x=0~2.0)材料电子结构与光学性质的第一性原理计算[J]. 发光学报, 2020, 41 (1): 38- 47. |
| 17 |
KORMANYOS A, THOMAS A, HUDA M N, et al. Solution combustion synthesis, characterization, and photoelectrochemistry of CuNb2O6 and ZnNb2O6 nanoparticles[J]. The Journal of Physical Chemistry C, 2016, 120 (29): 16024- 16034.
DOI |
| 18 |
WABURY M, MUELLER B H. ZnTa2O6, ein neuer vertreter des tri-α-PbO2 typs (mit ergä enzenden Daten über ZnNb2O6)[J]. Zeitschrift Für Anorganische und Allgemeine Chemie, 1984, 508 (1): 55- 60.
DOI |
| 19 |
刘东豪, 闫宇星, 沈静秋, 等. ZnNb2O6材料光电学特性研究[J]. 曲靖师范学院学报, 2019, 38 (6): 20- 25.
DOI |
| 20 |
HSIAO Y J, FANG T H, JI L W. Synthesis and luminescent properties of ZnNb2O6 nanocrystals for solar cell[J]. Materials Letters, 2010, 64 (23): 2563- 2565.
DOI |
| 21 | JAMES G S. Lange's handbook of chemistry[M]. 16th ed. New York: McGraw-Hill Companies Inc, 2005: 1.152-1.158. |
| 22 | MA R, ZHANG H L. Electronic properties of graphene nanoribbons doped with rhombus born nitride segment[J]. Chinese Journal of Computational Physics, 2019, 36 (1): 99- 105. |
| 23 | 高小奇, 郭志友, 张宇飞, 等. Al-N共掺杂ZnO电子结构和光学性质[J]. 发光学报, 2010, 31 (4): 509- 514. |
| 24 | 张梅玲, 陈玉红, 张材荣, 等. 内在缺陷与Cu掺杂共存对ZnO电磁光学性质影响的第一性原理研究[J]. 物理学报, 2019, 68 (8): 087101. |
| 25 | 沈学础. 半导体光谱和光学性质[M]. 北京: 科学出版社, 2002: 1- 32. |
| 26 | 黄昆. 固体物理学[M]. 北京: 高等教育出版社, 1988: 437- 462. |
| 27 | 李名復. 半导体物理[M]. 北京: 科学出版社, 2019: 161- 249. |
| 28 | 楼立人, 尹民, 李清庭. 发光物理基础: 固体光跃迁过程[M]. 合肥: 中国科技大学出版社, 2014: 35- 78. |
| 29 | ZHOU K, FENG Q, TIAN Y, et al. Oxidizing gas NO2 optical gas sensing characteristics of transition metal Cu and Cr doped TiO2 surfaces[J]. Chinese Journal of Computational Physics, 2018, 35 (6): 702- 710. |
| [1] | Yong FANG, Yongzhong JIN, Jian CHEN, Hongxiang ZONG, Liying ZHANG. Experimental and Simulation Studies on Relation Between Graphene Thickness and Its Force-distance Curve [J]. Chinese Journal of Computational Physics, 2021, 38(4): 441-446. |
| [2] | Jing PAN, Guohua SHEN. Enhanced Photocatalytic Activity of ZnO for Water-splitting with Isovalent Anion-Cation Codoping: First-principles Calculations [J]. Chinese Journal of Computational Physics, 2021, 38(3): 371-378. |
| [3] | ZHANG Le, SUN Bo, SONG Haifeng. First-principles Study of Hydrogen Behaviors in Plutonium Oxides [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(5): 595-602. |
| [4] | PENG Junhui. First-principles Study on Structures and Mechanical Properties of Ternary Layered Ceramics M-Al-N (M=Ti, Zr, Hf) [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(5): 603-611. |
| [5] | LI Lin, SUN Yuxuan, SUN Weifeng. Electronic Structure and Electrochromic Property of Sulvanite Compounds: A First-principles Study [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(4): 488-496. |
| [6] | WEN Shumin, YAO Shiwei, ZHAO Chunwang, WANG Xijun, LI Jijun. Effect of Strain on Electronic Structure and Optical Properties of Wurtzite GaN [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2020, 37(1): 119-126. |
| [7] | XIONG Zonggang, DU Juan, ZHANG Xianzhou. Acceptor and Donor Impurity States in Group V and VII Atom-doped Two-dimensional GeSe Monolayer [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(6): 733-741. |
| [8] | QIN Ping, GAO Zhenbang, LIU Haidi, CHEN Yingcai. First-principles Study of Transition Metal Monoboride TMB [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(4): 491-497. |
| [9] | LIU Huazhong, LUO Chunxia. First Principles Study of HCHO Adsorption on Hydroxylated TiO2-B(100) Surfaces [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2019, 36(3): 363-378. |
| [10] | XU Jian, DU Chengxu, DU Yingyan, JIA Qian, LIU Yanghua, WU Zhimin. First-principles Calculations of Magnetoelectric Properties of New Diluted Magnetic Semiconductor Mn-doped LiZnN [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2018, 35(6): 711-719. |
| [11] | CHEN Chuntian, CONG Shan, CHEN Hongfei, WANG Lei, LI Kai. First-Principles Study of Electronic Structure and Optical Properties of Bi Doped ZnO [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2018, 35(6): 720-728. |
| [12] | TIAN Zhuangzhuang, ZHOU Xiaoping, SONG Guolin. First-principles Calculation of Li Thin Films: Quantum Size Effects and Adsorption of Atomic Hydrogen [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2018, 35(6): 729-736. |
| [13] | TAN Junhua, PENG Junhui. First-Principles Study on Structure and Properties of Graphite Intercalation Compound HfC2 [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2018, 35(5): 613-618. |
| [14] | CAI Lugang. A First-Principles Study on Electronic and Optical Properties of Distorted Perovskite DyMnO3 [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2018, 35(3): 350-356. |
| [15] | WANG Wenhua, ZHAO Guojun, WANG Shudong. First-Principles Study of Thermoelectric Transport Properties of β-Antimonene [J]. CHINESE JOURNAL OF COMPUTATIONAL PHYSICS, 2018, 35(3): 365-372. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
Copyright © Chinese Journal of Computational Physics
E-mail: jswl@iapcm.ac.cn
Supported by Beijing Magtech Co., Ltd.