
计算物理 ›› 2022, Vol. 39 ›› Issue (5): 609-616.DOI: 10.19596/j.cnki.1001-246x.8483
收稿日期:2021-11-26
出版日期:2022-09-25
发布日期:2023-01-07
作者简介:罗强(1975-), 男, 硕士, 副教授, 研究方向凝聚态理论及纳米材料的计算机模拟,E-mail: 93414722@qq.com
基金资助:
Qiang LUO( ), Zhiwei MA, Guanzhen JIANG, Jiangfeng ZOU, Yi QIU
), Zhiwei MA, Guanzhen JIANG, Jiangfeng ZOU, Yi QIU
Received:2021-11-26
Online:2022-09-25
Published:2023-01-07
摘要:
基于密度泛函理论(DFT)的第一性原理方法, 用Ge原子分别替代纤锌矿AlN超晶胞中Al、N原子, 得到其结构、电子及光学性质。结果表明: 掺杂后结构发生了明显的改变, 两种方式的掺杂都使得AlN的晶格常数、体积和沿c轴方向的键长增加, 且晶格常数变化满足维加德(Vegard)定理; Ge原子替换Al原子形成能为5.41eV, Ge原子替换N原子形成能为5.58eV, 两种掺杂方式体系稳定性都低于纯AlN; 掺杂并未引入磁性, 前者引入施主杂质能带, 且杂质能带进入价带, 变为负能隙金属, 后者引入受主杂质能带, 禁带宽度为0.910eV, 两者均远小于本征AlN的禁带宽度4.040eV, 前者导电性显著提高, 后者导电性可能提高; 研究发现两种方式掺杂复折射率函数的虚部在低能区都近似不再为0, 表明两种掺杂都加强了AlN材料对低频电磁波的吸收能力; 前者介电函数虚部在低能区出现了新的波峰, 长波吸收能力更强, 后者在低能区未出现明显波峰, 能量损失均减少。
罗强, 马智炜, 蒋冠臻, 邹江峰, 邱毅. Ge掺杂AlN电子和光学性质的第一性原理计算[J]. 计算物理, 2022, 39(5): 609-616.
Qiang LUO, Zhiwei MA, Guanzhen JIANG, Jiangfeng ZOU, Yi QIU. First-Principles Calculations of Electronic and Optical Properties of Ge Doped AlN[J]. Chinese Journal of Computational Physics, 2022, 39(5): 609-616.
图1 AlN晶体结构 (a) 顶视图;(b) 侧视图
Fig.1 AlN crystal structure (a) top view; (b) side view
图2 Ge掺杂AlN的结构模型 (a) Ge替代Al;(b) Ge替代N
Fig.2 Structural models of Ge doped AlN (a) Ge instead of Al; (b) Ge instead of N
| 截断能Ec/eV | K网格点 | a(b)/nm | c/nm | 带隙值Eg/eV | 总能量E/eV |
| 460 | 4 × 4 × 3 | 0.626 | 1.003 | 4.014 | -6 240.343 |
| 460 | 5 × 5 × 4 | 0.626 | 1.003 | 4.014 | -6 240.354 |
| 517 | 5 × 5 × 4 | 0.626 | 1.003 | 4.040 | -6 242.769 |
| 550 | 5 × 5 × 4 | 0.626 | 1.003 | 4.045 | -6 243.425 |
| 550 | 6 × 6 × 5 | 0.626 | 1.003 | 4.045 | -6 243.425 |
表1 收敛性测试
Table 1 Convergence test
| 截断能Ec/eV | K网格点 | a(b)/nm | c/nm | 带隙值Eg/eV | 总能量E/eV |
| 460 | 4 × 4 × 3 | 0.626 | 1.003 | 4.014 | -6 240.343 |
| 460 | 5 × 5 × 4 | 0.626 | 1.003 | 4.014 | -6 240.354 |
| 517 | 5 × 5 × 4 | 0.626 | 1.003 | 4.040 | -6 242.769 |
| 550 | 5 × 5 × 4 | 0.626 | 1.003 | 4.045 | -6 243.425 |
| 550 | 6 × 6 × 5 | 0.626 | 1.003 | 4.045 | -6 243.425 |
| a(b)/nm | c/nm | 体积V/nm3 | 被替换位置沿c轴键长/nm | 被替换位置沿其他方向键长/nm | 带隙值Eg/eV | 总能量E/eV | |
| AlN | 0.622[ | 0.978[ | 0.379[ | 4.113[ | |||
| AlN | 0.626 | 1.003 | 0.393 | 0.191 | 0.190 | 4.040 | - 6 242.769 |
| Ge-AlN(Al) | 0.626 | 1.003 | 0.393 | 0.208 | 0.198 | 负 | - 8 661.174 |
| Ge-AlN(N) | 0.626 | 1.003 | 0.393 | 0.222 | 0.211 | 0.910 | - 8 491.680 |
表2 结构参数
Table 2 Structural parameters
| a(b)/nm | c/nm | 体积V/nm3 | 被替换位置沿c轴键长/nm | 被替换位置沿其他方向键长/nm | 带隙值Eg/eV | 总能量E/eV | |
| AlN | 0.622[ | 0.978[ | 0.379[ | 4.113[ | |||
| AlN | 0.626 | 1.003 | 0.393 | 0.191 | 0.190 | 4.040 | - 6 242.769 |
| Ge-AlN(Al) | 0.626 | 1.003 | 0.393 | 0.208 | 0.198 | 负 | - 8 661.174 |
| Ge-AlN(N) | 0.626 | 1.003 | 0.393 | 0.222 | 0.211 | 0.910 | - 8 491.680 |

图3 AlN电子性质 (a) 超晶胞能带结构;(b) 整体电子分波态密度(能量0处为费米能级);(c) Al原子电子分波态密度;(d) N原子电子分波态密度
Fig.3 AlN electronic properties (a) band structure of supercell; (b) global electron fractional density; (c) electron fractional density of Al atom; (d) electron fractional density of N atom
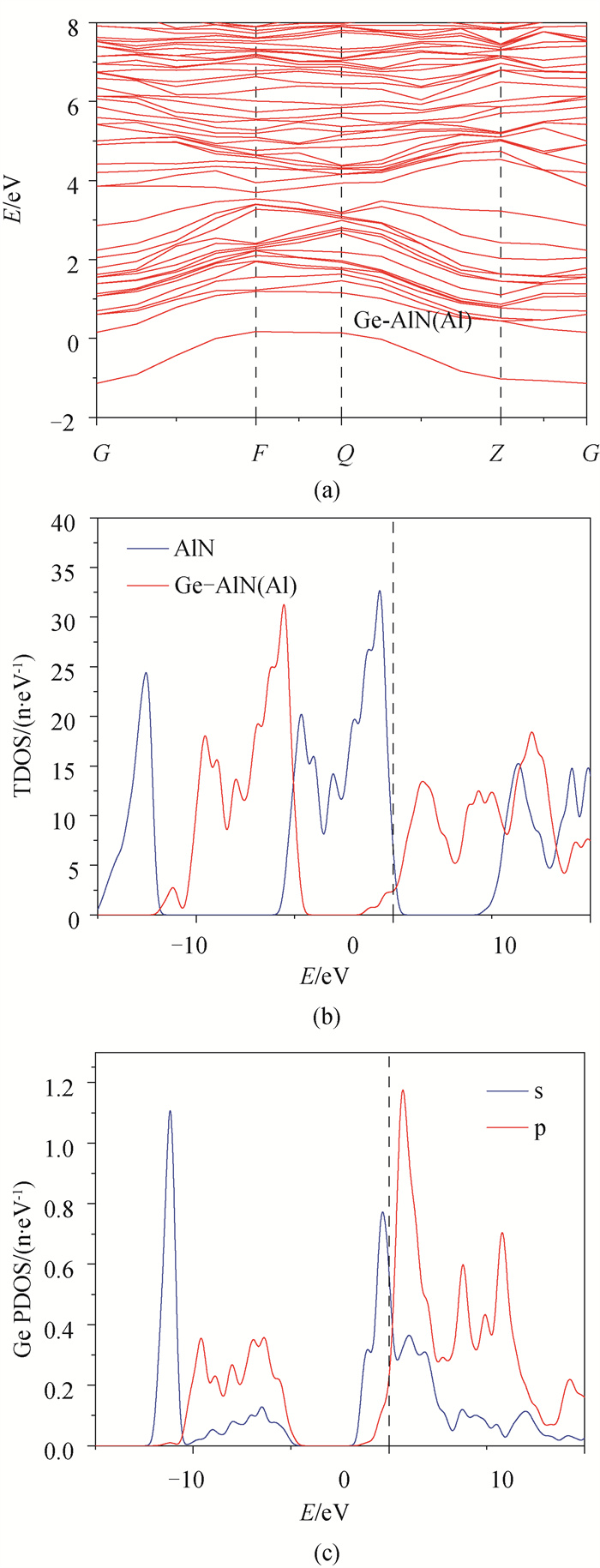
图4 (a) Ge-AlN(Al)超晶胞能带结构;(b) AlN与Ge-AlN(Al)总态密度;(c) Ge原子分波态密度
Fig.4 (a) Ge-AlN(Al) band structure of supercell; (b) AlN and Ge-AlN (Al) total state density; (c) electron fractional density of Ge atom
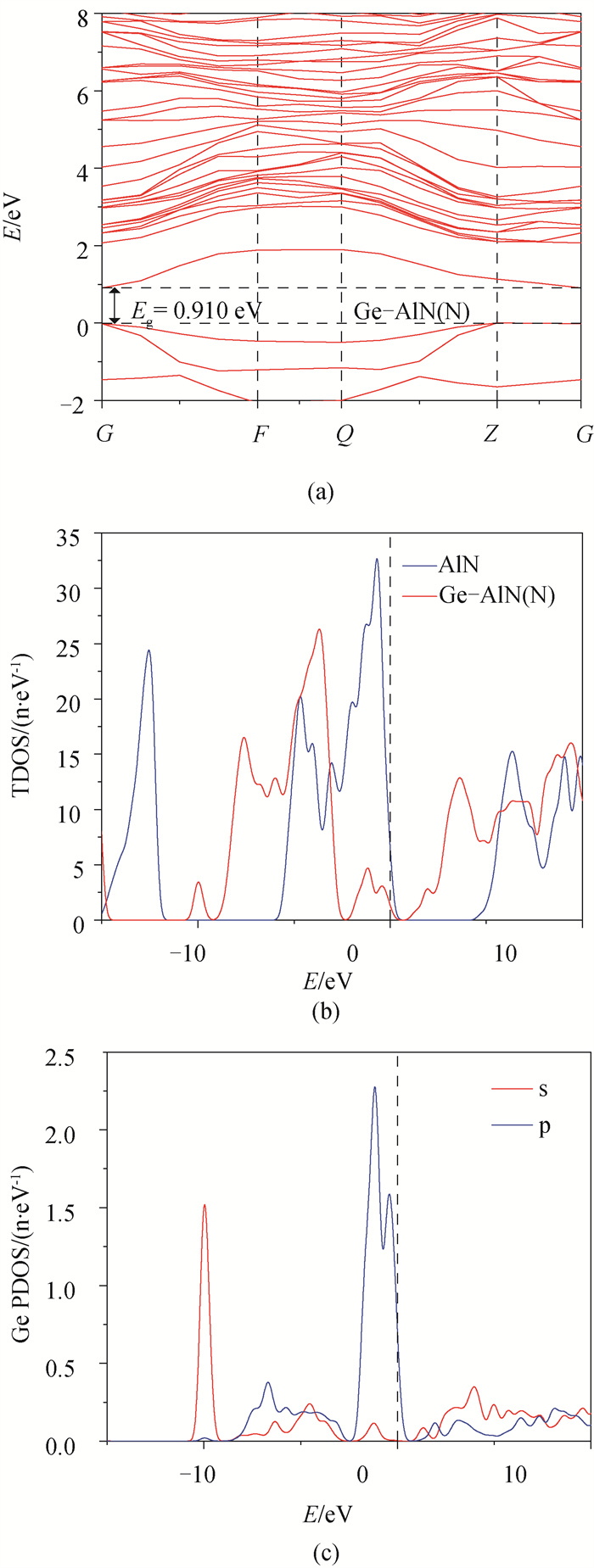
图5 (a) Ge-AlN(N)超晶胞能带结构;(b) AlN与Ge-AlN(N)总态密度;(c) Ge原子分波态密度
Fig.5 (a) Ge-AlN(N) band structure of supercell; (b) fractional density of AlN and Ge-AlN(N) electrons exclude doping position; (c) electron fractional density of Ge atom
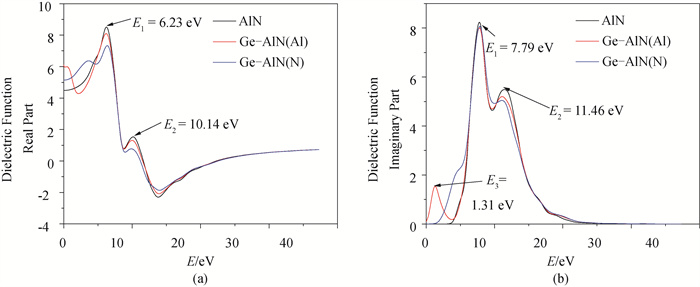
图6 介电函数 (a) 实部;(b) 虚部
Fig.6 Dielectric functions (a) real part; (b) imaginary part
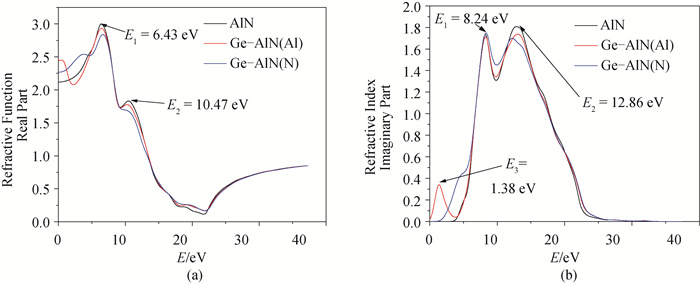
图7 复折射率函数 (a) 实部;(b) 虚部
Fig.7 Complex refractive index functions (a) real part; (b) imaginary part
| 1 |
DOI |
| 2 |
DOI |
| 3 |
DOI |
| 4 |
DOI |
| 5 |
DOI |
| 6 |
DOI |
| 7 |
DOI |
| 8 |
DOI |
| 9 |
DOI |
| 10 |
DOI |
| 11 |
DOI |
| 12 |
|
| 13 |
DOI |
| 14 |
DOI |
| 15 |
DOI |
| 16 |
张勇. 掺杂AlN的理论与实验研究[D]. 武汉: 华中科技大学, 2008.
|
| 17 |
|
| 18 |
DOI |
| 19 |
DOI |
| 20 |
DOI |
| 21 |
DOI |
| 22 |
DOI |
| 23 |
DOI |
| 24 |
DOI |
| 25 |
DOI |
| [1] | 严深浪, 项少辉, 龙孟秋. 锯齿型石墨烯纳米带结的自旋输运性质[J]. 计算物理, 2022, 39(6): 751-756. |
| [2] | 王晓慧, 张平. 高压下FCC相金属氢结构稳定性和非谐效应的理论研究[J]. 计算物理, 2022, 39(2): 159-164. |
| [3] | 房勇, 金永中, 陈建, 宗洪祥, 张丽英. 石墨烯厚度与其力-距离关系的实验和模拟研究[J]. 计算物理, 2021, 38(4): 441-446. |
| [4] | 闫宇星, 张珏璇, 郑帅, 汪帆, 熊琳强. 间隙原子对ZnNb2O6光电特性影响的第一性原理研究[J]. 计算物理, 2021, 38(4): 447-455. |
| [5] | 潘靖, 沈国华. 等价阴-阳离子共掺杂调节ZnO的能带结构及其光催化活性[J]. 计算物理, 2021, 38(3): 371-378. |
| [6] | 刘涛, 杨子义, 陈雨青, 高涛. 重费米子超导PuMGa5(M=Co,Rh)结构、电子和热力学性质的第一性原理研究[J]. 计算物理, 2021, 38(1): 106-112. |
| [7] | 张乐, 孙博, 宋海峰. 钚氧化物中氢行为的第一性原理研究[J]. 计算物理, 2020, 37(5): 595-602. |
| [8] | 彭军辉. 三元层状陶瓷M-Al-N(M=Ti,Zr,Hf)的结构及力学性质的第一性原理模拟[J]. 计算物理, 2020, 37(5): 603-611. |
| [9] | 赵一程, 郭俊宏, 胡芳仁. 应变对单层砷烯结构拉曼散射的影响[J]. 计算物理, 2020, 37(3): 365-370. |
| [10] | 温淑敏, 姚世伟, 赵春旺, 王细军, 李继军. 应变对纤锌矿结构GaN电子结构及光学性质的影响[J]. 计算物理, 2020, 37(1): 119-126. |
| [11] | 熊宗刚, 杜娟, 张现周. 二维GeSe纳米片五族和七族原子掺杂的受主和施主杂质态[J]. 计算物理, 2019, 36(6): 733-741. |
| [12] | 秦平, 高振帮, 刘海敌, 陈英才. 过渡金属单硼化物TMB的第一性原理研究[J]. 计算物理, 2019, 36(4): 491-497. |
| [13] | 刘华忠, 罗春霞. 甲醛分子在羟基化TiO2-B(100)面吸附的第一性原理研究[J]. 计算物理, 2019, 36(3): 363-378. |
| [14] | 周康, 冯庆, 田芸, 李科, 周清斌. 过渡金属Cu、Cr掺杂TiO2表面氧化性气体NO2光学气敏传感特性[J]. 计算物理, 2018, 35(6): 702-710. |
| [15] | 徐建, 杜成旭, 杜颖妍, 贾倩, 刘洋华, 毋志民. Mn掺杂LiZnN新型稀磁半导体磁电性质的第一性原理计算[J]. 计算物理, 2018, 35(6): 711-719. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
版权所有 © 《计算物理》编辑部
地址:北京市海淀区丰豪东路2号 邮编:100094 E-mail:jswl@iapcm.ac.cn
本系统由北京玛格泰克科技发展有限公司设计开发